


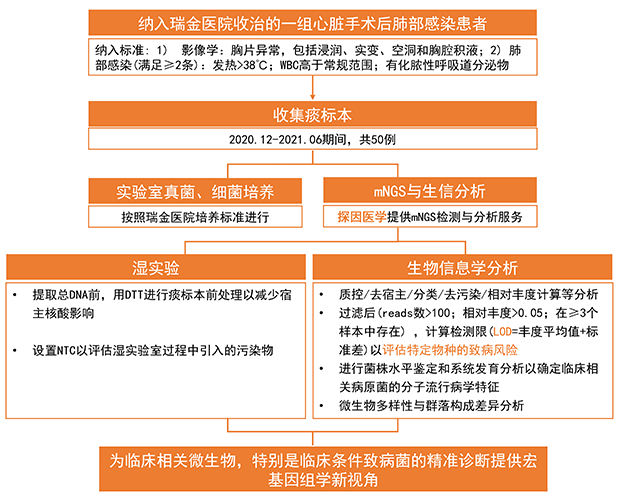
2022年3月3日,探因医学联合上海交通大学医学院附属瑞金医院心血管外科在Frontiers in Cellular and Infection Microbiology (IF: 5.293) 期刊发表研究型文章。瑞金医院顾坚未教授担任通讯作者,瑞金医院陈俊佶医师、探因医学孙连杰为共同第一作者。研究提出的“病原风险检测限”可以减少定植菌在特定身体部位的干扰,有利于临床解读mNGS结果;同时,也为评估复杂感染性疾病中多种微生物的致病风险提供了有力的参考模型。

· 文 / 章 / 要 / 点
人体呼吸道微生物群落中存在许多定植的条件致病菌,当微生物群落动态平衡被打破时,可能会引起感染。尽管宏基因组下一代测序(mNGS)技术具有更高的灵敏度和分辨率,有助于更好地了解痰标本中的微生物构成,但迄今为止,如何区分条件致病菌的定植或感染仍是临床病原体诊断的一个关键问题。
基于此背景,本研究对2020年12月至2021年6月间收治于瑞金医院心血管外科的50例心脏手术后肺部感染患者的痰标本常规培养结果和宏基因组测序(mNGS)数据进行了回顾性分析。揭示了痰微生物群落多样性,构建了评估单个微生物病原风险检测限(LOD),通过系统发育分析确定了临床相关病原菌的分子流行病学特征。
结果表明,根据本研究提出的基于mNGS检测结果的病原风险筛选策略预测的候选致病菌不仅与培养结果高度匹配,还提供了培养所遗漏的额外的重要病原体,为临床微生物学诊断提供了补充证据。研究结果为临床相关微生物,特别是临床条件致病菌的精准诊断提供了一种宏基因组学新视角。
· 研 / 究 / 内 / 容
· 主 / 要 / 结 / 果
| 物种鉴定与致病风险分析
通过培养法在所有样本中共鉴定出12种细菌、2种真菌,56%的样本(n=28)未通过培养法检出任何病原(表1)。相反,通过mNGS检出大量微生物,包括682种细菌、58种真菌和21种病毒。在个别样本中,mNGS检出的细菌和真菌种类数远远高于培养。此外,通过mNGS在其中31例(62%)样本中检出了病毒(表1)。
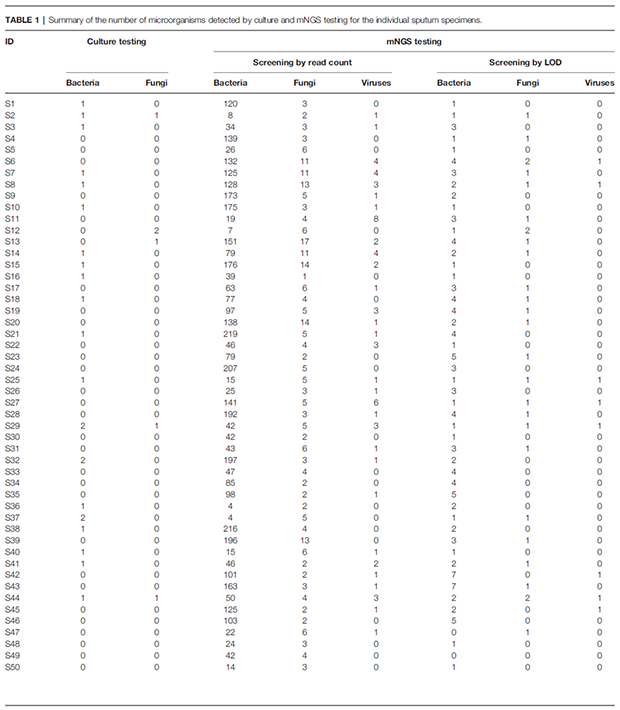
表1:痰标本培养和mNGS检出的微生物种类数总结
为评估单个微生物的致病风险,基于mNGS数据分别计算细菌、真菌和病毒的物种水平丰度LOD以筛选候选病原体。通过培养法检出的大多数细菌和真菌都包括在筛选出的病原体列表中(表2)。例如,2号患者培养和mNGS均检出新洋葱伯克氏菌和白色念珠菌。通过mNGS在44号患者中检出纹带棒状杆菌(56.2%), 金黄色葡萄球菌(39.2%), 白色念珠菌(93.1%), 黄曲霉(5.5%), 和人疱疹病毒1(99.8%)等5种病原体,培养也检出了金黄色葡萄球菌和白色念珠菌。
根据肺炎克雷伯菌的LOD(30.8%),其被评估为1(85.4%)、4(87.1%)、16(97.7%)、40(72.1%)号患者的病原体,除4号患者外,其余患者痰培养均为肺炎克雷伯菌阳性。患者4的mNGS结果确定肺炎克雷伯菌(87.1%)和白色念珠菌(97.8%)为病原体,但培养均未检出。总的来说,根据本研究提出的mNGS筛选标准预测的候选致病菌不仅与培养结果高度匹配,还提供了培养所遗漏的额外的重要病原体,为临床微生物学诊断提供了补充证据。
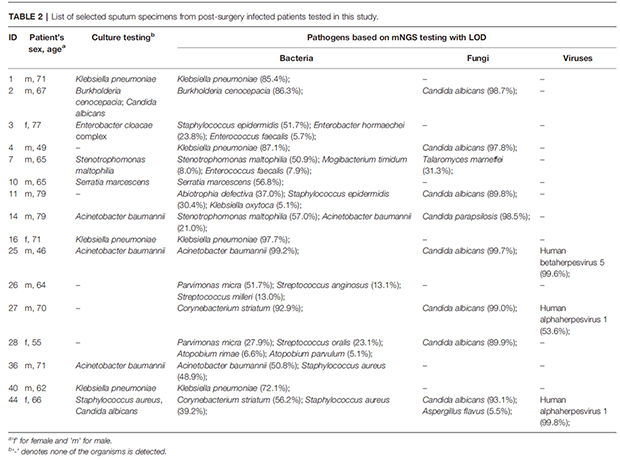
表2:在本研究中测试的术后感染患者痰标本列表
| 痰微生物组的群落结构
mNGS数据分析结果表明,细菌群落多样性高于真菌和病毒,因此,主要聚焦于细菌群落进行多样性分析。物种积累曲线趋于平滑,表明细菌物种的数量随着样本量的增加而缓慢变化(图1A)。如图1B所示,痰标本中丰度平均值排在前五位的细菌物种分别是肺炎克雷伯菌(7.3%)、嗜麦芽窄食单孢菌(5.6%)、表皮葡萄球菌(4.9%)、婴儿链球菌(4.8%)和鲍曼不动杆菌(3.6%)。此外,部分病原体在一些样本中占显著主导地位,例如1、4、16、40号样本中的肺炎克雷伯菌,25、36号样本中的鲍曼不动杆菌,9、17、46号样本中的浅黄奈瑟菌。
研究者进一步比较了两种临床结果(恢复 vs. 恶化)之间痰微生物组的群落结构动态差异,α多样性分析结果表明,两组间的群落多样性差异不显著(p = 0.125)(图 1C)。ANOSIM分析结果表明,组间差异显著低于恶化组的组内差异 (R = 0.173, p = 0.008) (图 1D)。同时,低R值表明临床结果对微生物群落的影响较弱。α和β多样性分析结果共同表明,细菌群落多样性与术后感染患者临床结果的相关性较小,可能是由于不同病原体在个体样本中占主导地位所致。
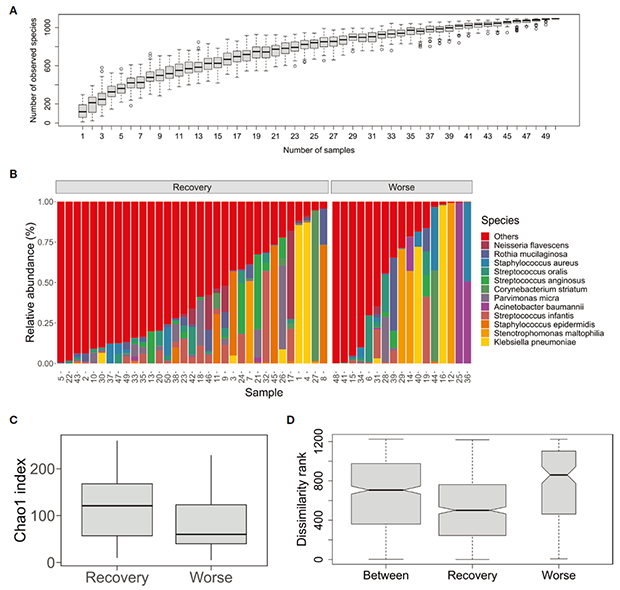
图1:术后肺部感染患者痰微生物群落结构及多样性
| 病原体的菌株水平特征
菌株鉴定对于更好地了解微生物致病性和临床相关微生物的传播具有重要意义。根据 mNGS测试评估的病原体列表,对肺炎克雷伯菌、纹带棒状杆菌、金黄色葡萄球菌和白色念珠菌这4种条件致病菌进行了菌株水平分析。菌株水平系统发育树展示了从本研究宏基因组数据中获得的菌株序列与具有公开基因组序列的临床微生物分离物之间的相关性(图 2)。
进一步分析表明,来自4号患者的肺炎克雷伯菌菌株为 ST29型,而分别来自1、16号患者的两株可能为新的 ST 型。其中,4号患者的菌株与从尿路感染中分离的多重耐药ST29菌株(CP024489/INF249)关系最密切,16号患者的菌株与引起肝脓肿和脑膜炎的肺炎克雷伯菌菌株NTUHK2044(AP006725)密切相关(图2A)。
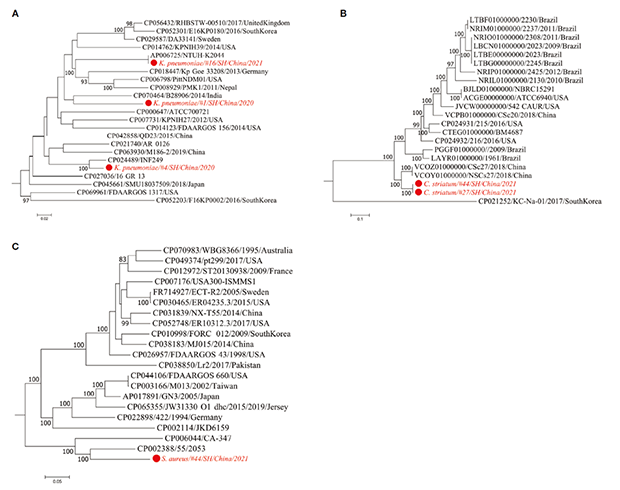
图2:三种细菌病原体的最大似然法系统发育树
来自27、44号患者的两个纹带棒状杆菌菌株与继发性下呼吸道细菌感染的流感患者中分离出的两个菌株(VCOZ00000000和VCOY00000000)关系密切(图 2B)。此外,44号患者中的金黄色葡萄球菌菌株与导致20世纪50年代产PVL金黄色葡萄球菌全球大流行的55/2053菌株密切相关(图 2C)。
图3展示了分别基于StrainPhlAn和MLST方法进行的白色念珠菌系统发育分析结果,两种方法均表明来自20号患者的白色念珠菌菌株与CHN1、Ca529L、A123和A67菌株密切相关。
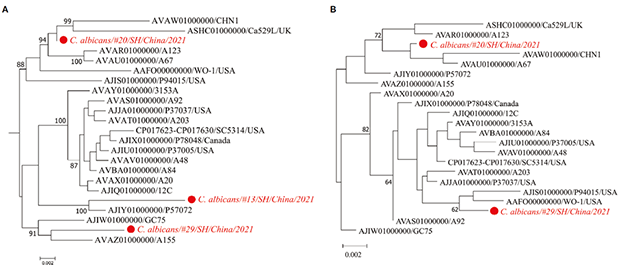
图3:白色念珠菌的最大似然法系统发育树
· 讨 / 论
呼吸道等特定身体部位微生物群落的平衡与宿主健康和疾病密切相关。疾病状态下,微生物群失衡可改变某些条件致病菌株的比例,从而促进由这些菌株主导的复杂群落增殖并进一步引起疾病感染。预测条件致病菌的感染风险对这些临床相关菌株的诊断具有重要意义。因此,本研究探索了基于mNGS物种丰度谱的筛选标准,进一步提出LOD以评估本研究中痰微生物组的致病风险。
估计微生物组中不同微生物的致病风险有助于区分临床标本中的致病菌株和定植菌株。本研究通过基于mNGS数据的筛选标准获得的病原体清单几乎涵盖了所有培养鉴定出的物种,并揭示了一些感染相关的其他病原体。
痰微生物组中存在许多定植菌,也是条件致病菌。如,革兰氏阳性菌肺炎链球菌是鼻咽部常见共生菌之一,也是一种可导致肺炎、菌血症、脑膜炎等的机会性致病菌。本研究中超过一半样本检出了肺炎链球菌,基于LOD(3.6%)被确定为6、33、42号患者的病原体;粘质沙雷氏菌是一种革兰氏阴性条件医院感染病原体,在10号患者中被检测为单一病原体,与培养结果相一致;白色念珠菌在正常情况下是一种无害的共生酵母,也是一种可引起粘膜症状性感染的条件致病菌。本研究中,白色念珠菌在约一半的样本中均有检出,并被进一步评估为30%样本的高风险病原真菌。这表明,对于条件致病菌引起的感染来说,LOD值可能是一个有希望的临床指标,但仍需更大的样本量为LOD的统计估计提供可靠的微生物丰度参考。
挖掘宏基因组数据可获得菌株水平的更准确的分类信息,可进一步用于解释新检出菌株的致病机制、耐药性和传播。例如,尽管4号患者痰培养显示肺炎克雷伯菌阴性,mNGS在该患者中检出了肺炎克雷伯菌,进一步分析表明这种高丰度的肺炎克雷伯菌菌株与多重耐药、高毒力分离株INF249在系统发育上相关,而16号患者中检出的肺炎克雷伯菌菌株与NTUH-K2044这种K1型、ST23型的高毒力菌株密切相关。此外,13号患者中检出的白色念珠菌菌株与血流感染相关白色念珠菌P57072菌株的关系最为密切。由于条件致病菌通常包括致病菌株和共生菌株,菌株水平系统发育是提供准确检测其是否为致病菌株的替代方法。
· 文 / 章 / 总 / 结
本研究基于mNGS物种丰度数据提出了一种可用于评估肺部感染患者痰微生物致病风险的实用筛查策略。该策略能减少定植微生物的干扰,也可以区分致病菌株和特定身体部位的定植菌株。本研究为临床相关微生物,尤其是临床环境中的条件致病菌的精确诊断提供了一种宏基因组学新见解。



